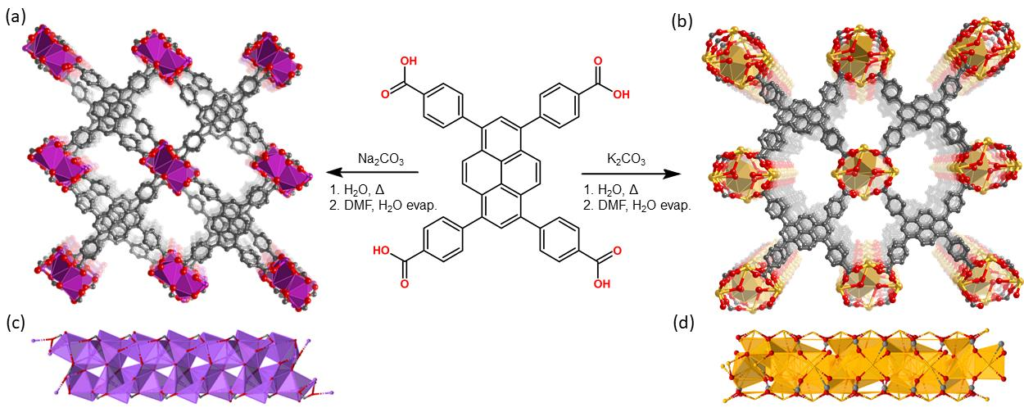
I’m apparently now an expert on metal organic frameworks (MOFs) with the publication of this article in CrystEngComm, DOI: 10.1039/D0CE01505A. In reality, I only worked on the spectroscopy sections — congratulations to Chris who actually touched the chemicals!
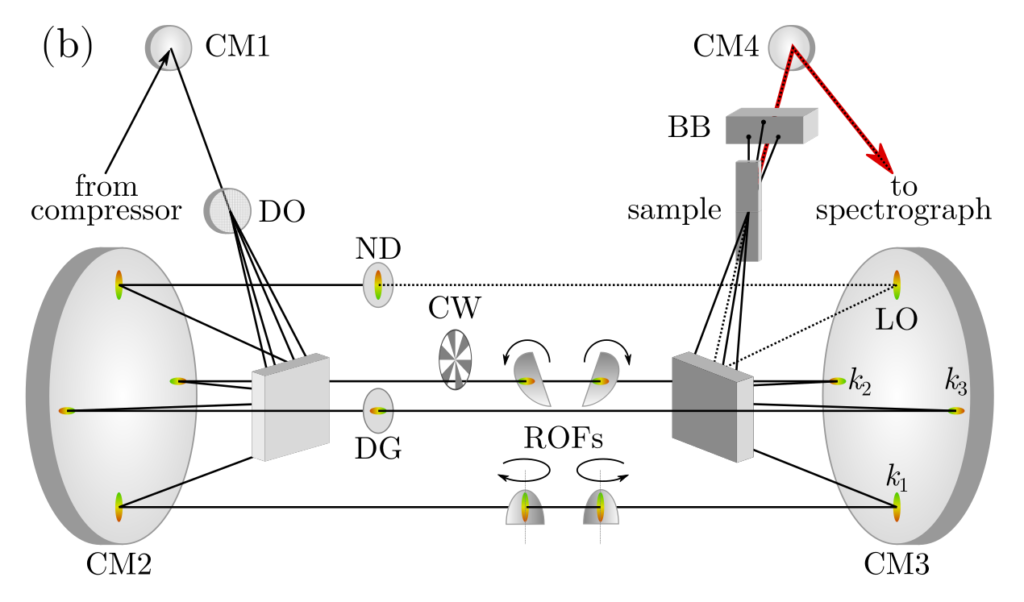
Paper describing the 2D spectrometer design was published in The Journal of Physical Chemistry A (DOI: 10.1021/acs.jpca.0c00285). The accepted version of the manuscript can be downloaded here, along with the supporting information.
Layout of the 2D spectrometer. CM: concave mirrors, DO: 2-dimensional diffractive optic, ND: neutral density filter, DG: delay glass, CW: chopper wheel, ROFs: rotating optical flats, BB: beam block. The positions of the laser pulses are labeled with kn and LO. The third-order signal (red line) is emitted from the sample on the same path as the LO (dotted line).
This paper was a pure computational chemistry project studying the photoexcited state of a common conjugated polymer used in organic photovoltaics. It was published in the Journal of Physical Chemistry Letters, DOI: 10.1021/acs.jpclett.7b01053.
This was one of those “why hasn’t anyone done this yet?” projects which Tak gave to one of his honours students, Ras Roseli. She did a great job on the computational work, which I can claim no credit for! I provided the small amount of experimental data and wrote up the manuscript with Tak.
While I think this is a great piece of work for a (technically undergraduate) student, the field of computational chemistry moves fast. If you’re looking to replicate the techniques in this work then I’d recommend looking at some of the newer hybrid density functionals. For example, ωB97X-D, or the even newer ωB2PLYP developed by the Goerigk group.
Fun fact: There’s a spelling error on the first page which is entirely my fault. See if you can find it.
Here’s my Ph.D thesis, entitled Theoretical and Spectroscopic Studies of Energy and Charge Transport in Organic Semiconductors. It was obtained at the University of Adelaide under the supervision of Tak W. Kee and David M. Huang.
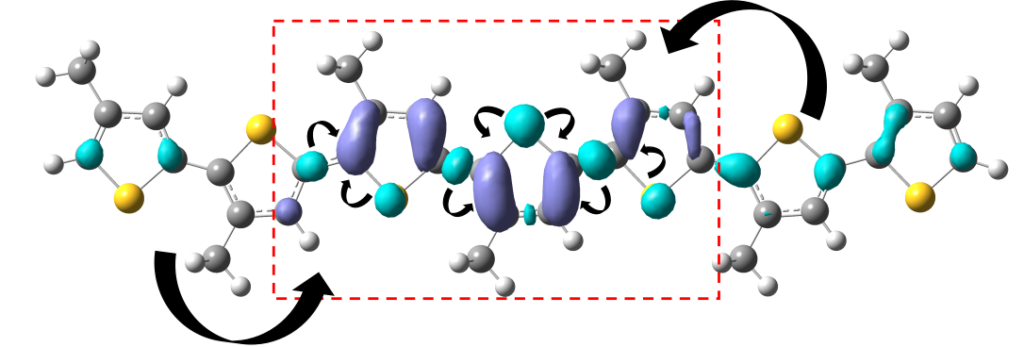
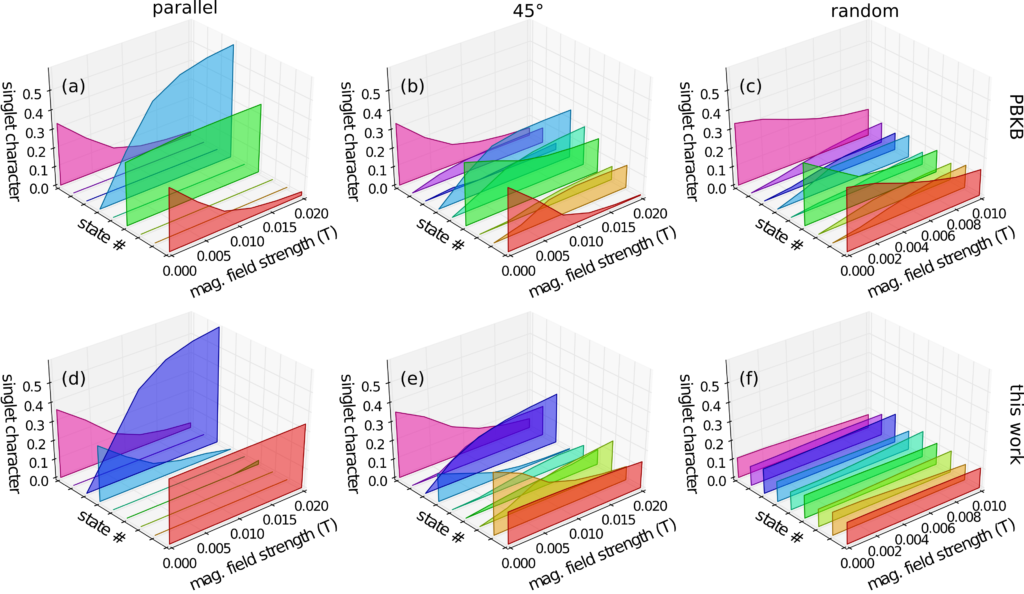
This was a purely computation project based around a quantum mechanical model of molecular spin interactions in the presence of magnetic fields, for the purpose of predicting efficiency of triplet-triplet annihilation upconversion and singlet fission. This was published in the Journal of Physical Chemistry C, DOI: acs.jpcc.6b04934.
It was written in the form of a Comment on a previously published paper. The reason for this was we wanted to use the spin-Hamiltonian method as part of a chapter in my PhD thesis, however the current state of the literature was, frankly, quite obviously wrong. Hence, we took a six month detour to first work on correcting the currently accepted literature reference on the molecular spin-Hamiltonian.
It actually felt kind of bad publishing this, since the Bardeen group is well known and respected. However, it’s science, and if it’s wrong, it’s wrong… right? Funnily enough, after this correction was published, Bardeen contacted David to say “no hard feelings”, which was nice. A few weeks later they published this article. My guess is they had been sitting on that experimental data but since it didn’t match their (wrong) calculations, they didn’t want to publish it. Once the model was corrected, their experimental results actually made sense!
There’s another funny story about this paper. We got sent someone’s manuscript for review which was almost a complete copy of this work. Except they made many of the same mistakes as in the original paper and were claiming we were actually wrong. What made it really funny was they had directly copied passages of text from our article and figure captions. Yes, that paper did eventually get published, and still has some of the original mistakes in it.
This project was primarily computational, involving the development of a hybrid classical and quantum method. It was published in the Journal of Physical Chemistry C, DOI: 10.1021/acs.jpcc.5b00705.
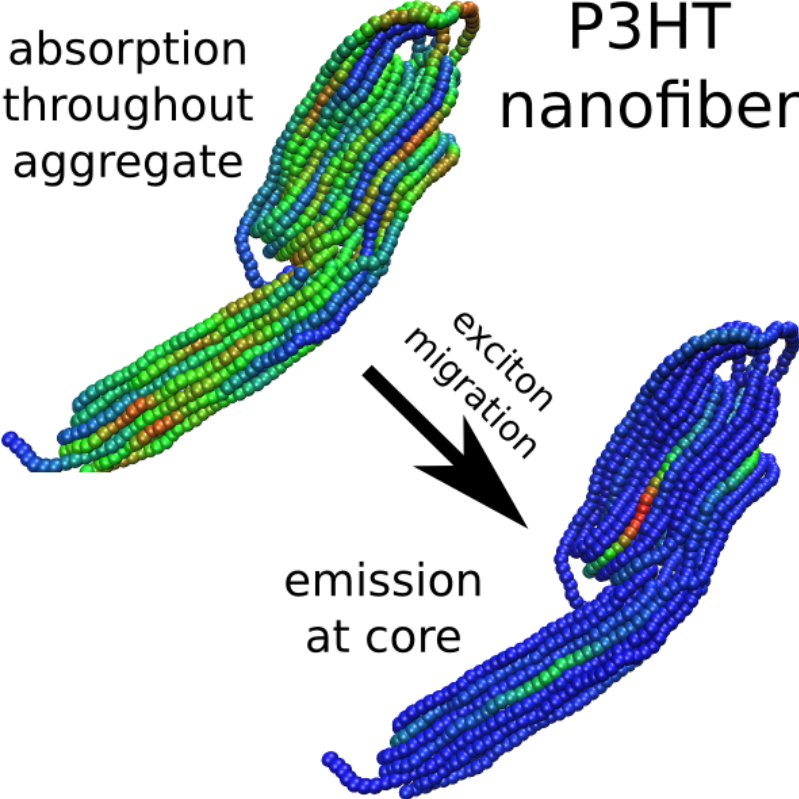
The morphology dependence of exciton transport in the widely used conjugated polymer poly(3-hexylthiophene) (P3HT) is elucidated by combining an accurate mesoscale coarse-grained molecular dynamics simulation model of P3HT structure with a Frenkel–Holstein exciton model. This model provides a more realistic representation than previously achieved of the molecular-level details of exciton transport on large length scales relevant to electronic applications. One hundred 300-monomer regioregular P3HT chains are simulated at room temperature for microseconds in two implicit solvents of differing solvent quality in which the polymer chains adopt contrasting morphologies: nanofiber-like aggregates or well-separated extended conformations. The model gives reasonable quantitative agreement with steady-state absorption and fluorescence and time-resolved fluorescence experiments, and provides valuable insight into the mechanism of exciton transport in conjugated polymers. In particular, exciton transfer in nanofiber aggregates is shown to occur mainly through interchain hops from chromophores on the aggregate surface toward the aggregate core, a behavior with important implications for organic electronic applications. Furthermore, the counterbalancing effects of greater orientational order and faster exciton transport in nanofiber aggregates than in extended chains are found to explain the puzzling observation of similar fluorescence anisotropy decay rates in nanofibers and free chains.
This was my first publication during my PhD. It focused on a three-pulse transient absorption technique applied to a common conjugated polymer material. It was published in the Journal of Physical Chemistry Letters, DOI: 10.1021/jz500217f.
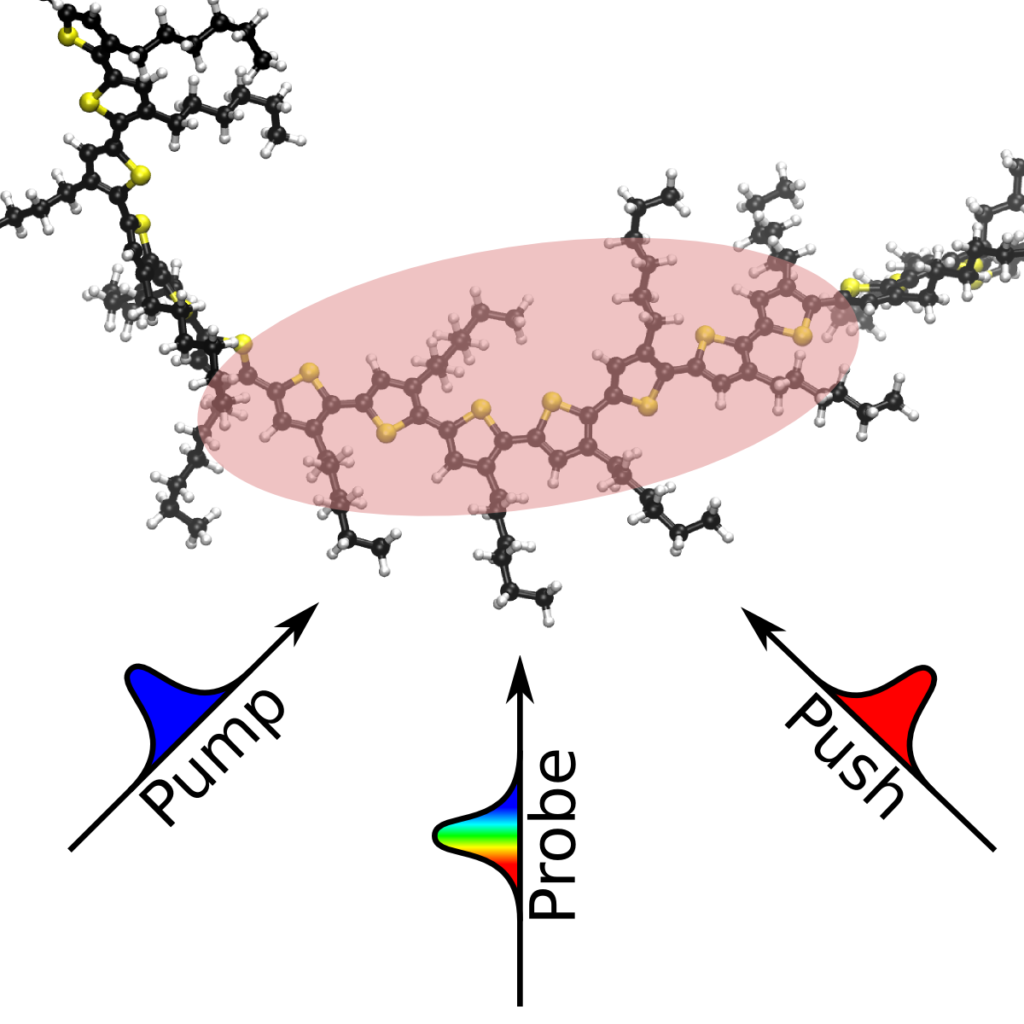
The dynamics of high-energy excitons of poly(3-hexylthiophene) (P3HT) are shown to consist of torsional relaxation and exciton dissociation to form free carriers. In this work, we use pump–push–probe femtosecond transient absorption spectroscopy to study the highly excited states of P3HT in solution. P3HT excitons are generated using a pump pulse (400 nm) and allowed to relax to the lowest-lying excited state before re-excitation using a push pulse (900 or 1200 nm), producing high-energy excitons that decay back to the original excited state with both subpicosecond (0.16 ps) and picosecond (2.4 ps) time constants. These dynamics are consistent with P3HT torsional relaxation, with the 0.16 ps time constant assigned to ultrafast inertial torsional relaxation. Additionally, the signal exhibits an incomplete recovery, indicating dissociation of high-energy excitons to form charge carriers due to excitation by the push pulse. Our analysis indicates that charge carriers are formed with a yield of 11%.