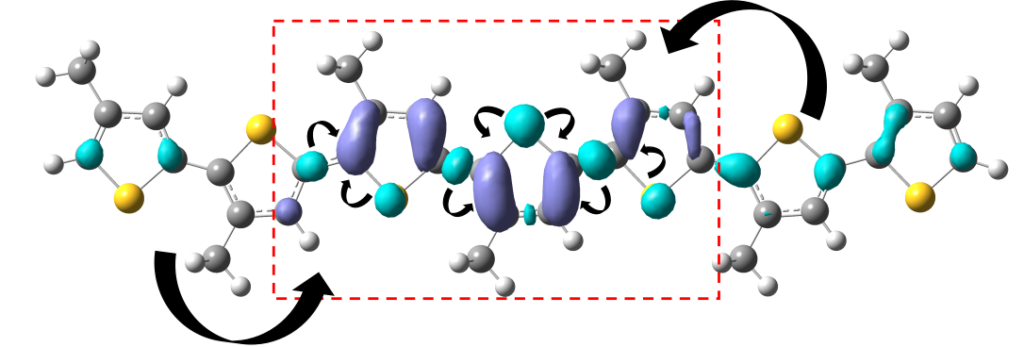
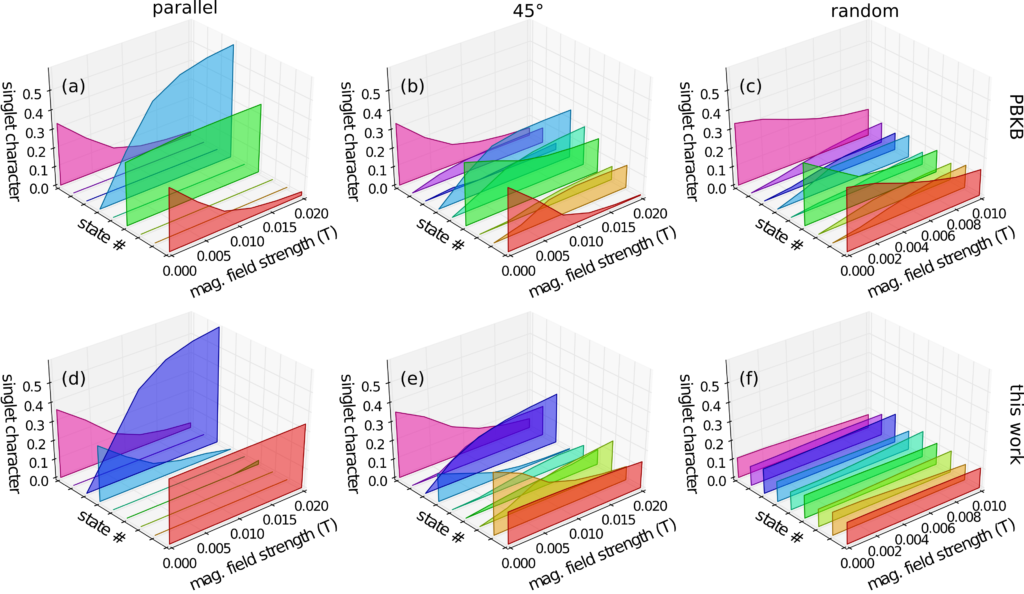
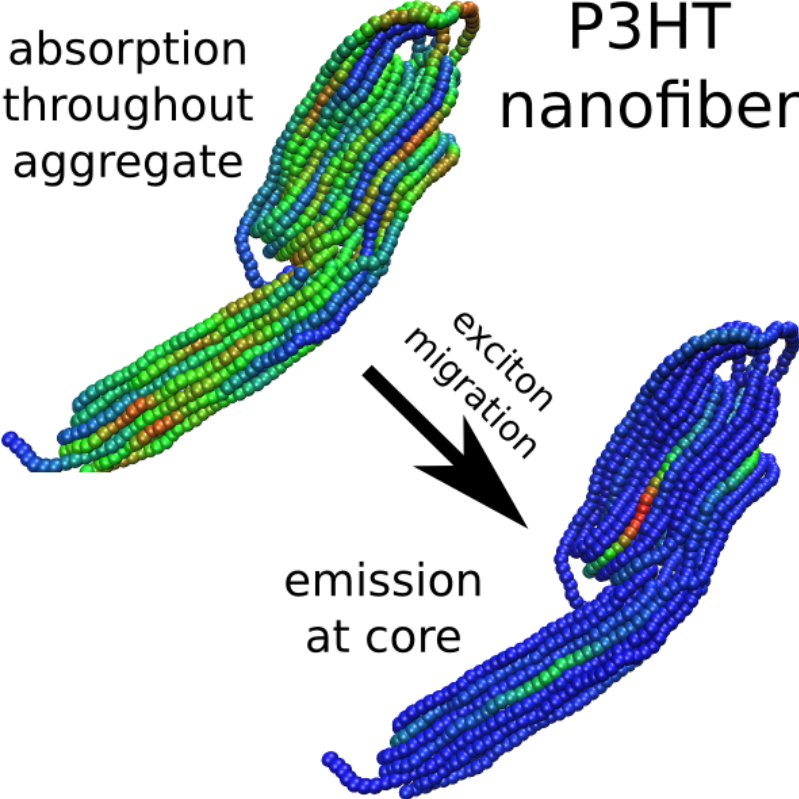
This paper was a pure computational chemistry project studying the photoexcited state of a common conjugated polymer used in organic photovoltaics. It was published in the Journal of Physical Chemistry Letters, DOI: 10.1021/acs.jpclett.7b01053.
This was one of those “why hasn’t anyone done this yet?” projects which Tak gave to one of his honours students, Ras Roseli. She did a great job on the computational work, which I can claim no credit for! I provided the small amount of experimental data and wrote up the manuscript with Tak.
While I think this is a great piece of work for a (technically undergraduate) student, the field of computational chemistry moves fast. If you’re looking to replicate the techniques in this work then I’d recommend looking at some of the newer hybrid density functionals. For example, ωB97X-D, or the even newer ωB2PLYP developed by the Goerigk group.
Fun fact: There’s a spelling error on the first page which is entirely my fault. See if you can find it.